In this blog post, I will give a short overview of our recent research paper about protein structure analysis and classification by my team (M. S. Nawaz, P. Fournier-Viger, Y. He and Q. Zhang).
Proteins are essential molecules for life, as they perform a variety of functions in living organisms, such as catalysis, transport, signaling, defense, and regulation. The structure of proteins determines their function and interactions with other molecules. Therefore, understanding the structure of proteins is crucial for advancing biological and medical research. In biophysics and computational biology, predicting 3D structure of protein from its amino acid sequence is an important research problem.
However, protein structure analysis and classification is a challenging task, as proteins have complex and diverse shapes that can be affected by various factors, such as environment, mutations, and interactions. Moreover, the number of protein structures available in databases such as PDB and EMDB is increasing rapidly, making it difficult to manually annotate and compare them.
Thus, we recently published a research paper, which proposes a novel method for protein structure analysis and classification. The paper is titled “PSAC-PDB: Analysis and Classification of Protein Structures” and it will appear in the journal Computers in Biology and Medicine, published by Elsevier. The reference of the paper is:
Nawaz, S. M., Fournier-Viger, P., He, Y., Zhang, Q. (2023). PSAC-PDB: Analysis and Classification of Protein Structures. Computers in Biology and Medicine, Elsevier, Volume 158, May 2023, 106814
The main contribution of the paper is to propose a new framework called PSAC-PDB (Protein Structure Analysis and Classification using Protein Data Bank).
The main contribution of the paper is to propose a new framework called PSAC-PDB. A sechma of the overall method can be found below:
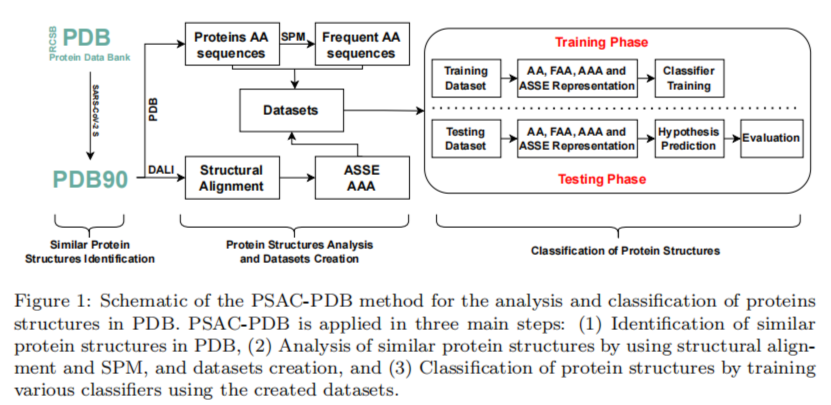
Briefly, PSAC-PDB is a computational method that uses a protein structure comparison tool (DALI) to find similar protein structures to a query structure in PDB, and then uses amino acid sequences, aligned amino acids, aligned secondary structure elements, and frequent amino acid patterns to perform classification. PSAC-PDB applies eleven classifiers and compares their performance using six evaluation metrics.
PSAC-PDB also uses sequential pattern mining (SPM) to discover frequent amino acid patterns that can improve the classification accuracy. SPM is a data mining technique that finds subsequences that appear frequently in a set of sequences. SPM can capture the sequential ordering and co-occurrence of amino acids in protein sequences, which can reflect their structural and functional properties. Some examples of frequent sequential patterns of Amino Acids (AA) extracted by the TKS and CM-SPAM algorithms are shown as example in the table below for three families: the S protein structure of SARS-CoV-2 (SSC2), the S protein structures of other viruses and organisms (SO), and Protein (enzyme) structures for others (O).

PSAC-PDB was tested on the case study of SARS-CoV-2 spike protein structures, which are responsible for the entry of the virus into host cells. PSAC-PDB finds 388 similar protein structures to the query structure in PDB, and divides them into three families: S protein structures of SARS-CoV-2, S protein structures of other viruses and organisms, and other protein structures. PSAC-PDB uses four datasets based on amino acid sequences, aligned amino acids, aligned secondary structure elements, and frequent amino acid patterns for classification.
Results have shown that PSAC-PDB achieves high accuracy, precision, recall, F1-score, MCC and AUC values for all three families of protein structures, than state-of-the-art approaches for genome sequence classification. Here are some of the results showing this:
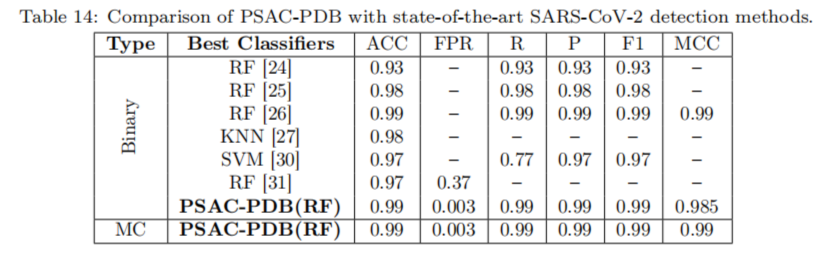
But what is also interesting is that PSAC-PDB shows that using frequent amino acid patterns or aligned amino acids can improve the classification performance compared to using only amino acid sequences or aligned secondary structure elements. Thus, PSAC-PDB can benefit the research community in structural biology and bioinformatics.
For more information, please see the paper. Besides, the datasets from this paper can also be found on Github: https://github.com/saqibdola/PSAC-PDB and implementations of sequential pattern mining algorithms used in the paper can be found in the SPMF data mining software.



